Introduction
Our body requires ATP, which is generated by reducing O2 to H2O through oxidative phosphorylation(OxPhos) in mitochondria (Jarmuszkiewicz, et al., 2015). During this process, a group of reactive metabolites including free radicals(FR)(e.g. O2●-, OH●) and non-radicals(e.g. H2O2) are generated. They are grouped as reactive oxygen species(ROS) (Rao, et al., 2011).
FR are groups of atoms orbited by one or more unpaired electrons. Apart from mitochondria, FR are also generated by lysosomes, peroxisomes, endoplasmic reticulum(ER), plasma membrane and from metabolized drugs/toxins (Rao, et al., 2011). The intensity of FR generation increases with age (Kumar, et al., 2012). Presence of unpaired electrons makes FR highly reactive to start a chain reaction of oxidations, which impair normal cellular activities. This may lead to cell injury and death. Though H2O2 is not an FR, it behaves like one and can generate OH● through Fenton reaction. OH● is the most reactive FR. To negate the function of FR, a defensive mechanism called antioxidants[superoxide dismutase(SOD), glutathione(GSH), catalase, etc.] are present in human body (Rao, et al., 2011).
When equilibrium between producing ROS and detoxifying reactive intermediates or repairing oxidative damages fails, the condition is called as oxidative stress(OxiStres) (Dias, et al., 2013). Increased ROS activity leads to a cascade of steps that damages macromolecules like proteins, lipids, and DNA, resulting in a pathological condition. It is an underlying mechanism in chronic and degenerative disorders (Rao, et al., 2011). An example is Parkinson’s disease[PD] (Hwang, 2013).
PD, an escalating neurodegenerative and movement disease (Roberts, et al., 2015) , has clinical manifestations like tremor, bradykinesia, muscular rigidity, and postural imbalance (Hwang, 2013). Aetiology can be environmental, genetic or idiopathic. The main pathology is the loss of dopaminergic neurons at substantia nigra pars compacta(a part of basal ganglia) and their fibres residing in the striatum ,and accumulation of Lewy bodies (Manoharan, et al., 2016).
Pathophysiology of Parkinson’s disease
Basal ganglia(BG), a part of the brain, is an interconnected system containing globus pallidus interna(GPi), striatum(caudate and putamen), substantia nigra pars compacta(SNc), subthalamic nucleus(STN), globus pallidus externa(GPe), and substantia nigra pars reticulata(SNr)(Figure1). They are involved in both motor and cognitive functions. Striatum and STN form input nuclei whereas GPi and GPe form output nuclei of BG. Their network is categorised into direct and indirect pathways (Nelson & Kreitzer, 2014). Role of direct pathway is to initiate and cause motor movements while that of indirect pathway is to inhibit motor movements.
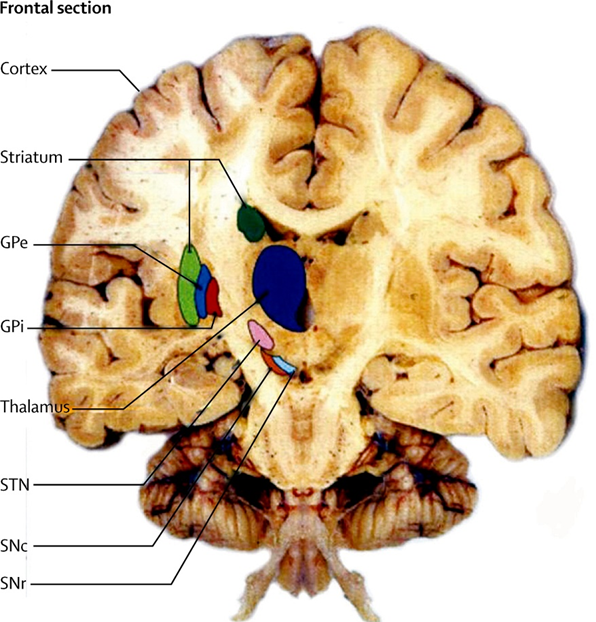
Figure1: Frontal section of brain showing the main components of basal ganglia. They are striatum(caudate and putamen), globus pallidus interna(GPi), globus pallidus externa(GPe), subthalamic nucleus(STN), substantia nigra pars compacta(SNc) and substantia nigra pars reticulata(SNr) (Obeso, et al., 2014).
In direct pathway, cortex sends excitatory(glutamate) signals to the striatum, whose excitation is amplified by acetylcholine and dopamine. Dopamine(excitatory) is signalled from SNc to bind to a D1 receptor of the striatum. The excited striatum then sends inhibitory(GABA) signals to SNr and GPi. The normal function of GPi and SNr is to inhibit thalamus. Since they get inhibited by the striatum, their inhibitory effect on thalamus weakens. This would then allow the thalamus to send excitatory(glutamate) signals to the cortex to cause movements(Figure2).
In indirect pathway, cortex sends excitatory(glutamate) signals to the striatum, whose excitation is amplified by acetylcholine. The striatum sends inhibitory(GABA) signals to GPe. This then inhibits GPe from sending inhibitory signals(GABA) to STN. Therefore, STN can excite(glutamate) GPi and SNr. GPi and SNr, upon excitation, send inhibitory(GABA) signals to thalamus to inhibit movements. Dopamine in this pathway inhibits the striatum by binding to its receptor D2. This allows GPe to inhibit STN, thereby aiding to increase movements (Nambu, 2009)(Figure2).
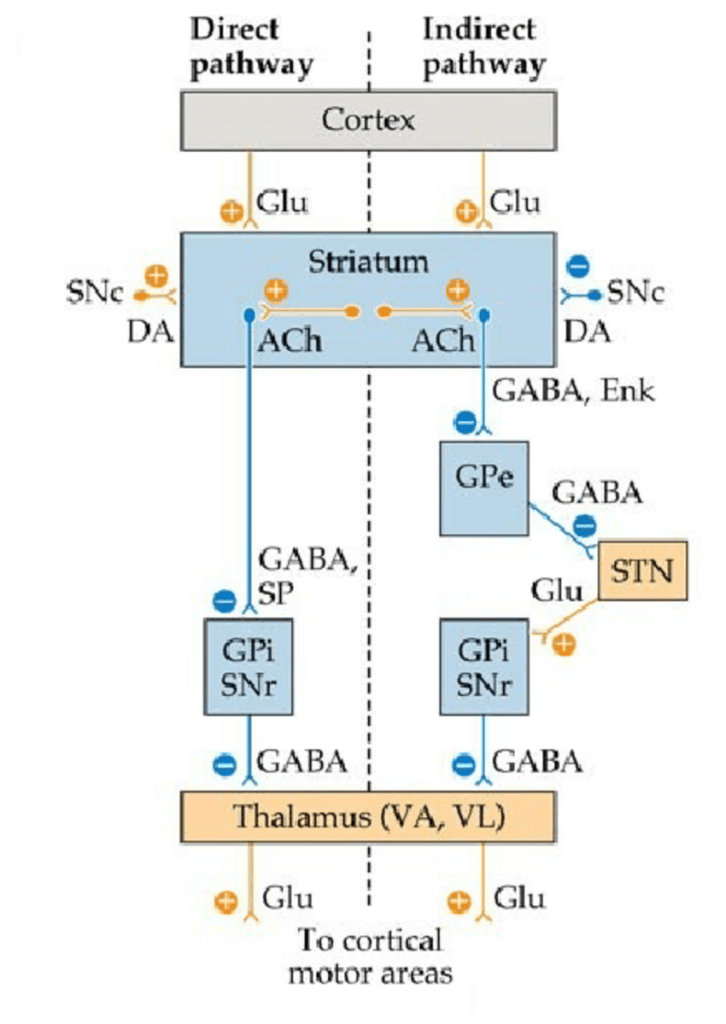
Figure2: Flowchart showing direct and indirect pathway of basal ganglia in a healthy brain. Direct pathway excites motor movements while indirect pathway inhibits motor movements. Dopamine in both the pathways aids in increased motor movement (Leisman, et al., 2013). (+ is excitatory signal, – is inhibitory signal, Glu is glutamate, DA is dopamine, Ach is acetylcholine, SNc is substantia nigra pars compacta, GPe is globus pallidus externa, STN is subthalamic nucleus, GPi is globus pallidus interna, and SNr is substantia nigra pars reticulata).
Dopamine allows increased movement by exciting direct pathway and inhibiting indirect pathway(Figure2). In patients with PD, dopamine is lost, hence, hypokinetic movements like bradykinesia are seen (Magrinelli,, et al., 2016). Hyperkinetic movements like tremors are also seen due to excitation of striatum by acetylcholine (Potkonjak & Cox, 1969 ).
One mechanism responsible for the loss of dopaminergic neurons in PD is OxiStres. Sources of OxiStres in PD are dopamine metabolism, dysfunctional mitochondria (Hwang, 2013), accumulation of iron, genetic mutations (Kumar, et al., 2012) and neuroinflammation (Hwang, 2013). Its consequences include oxidation of proteins and DNA, lipid peroxidation, and abnormal calcium influx (Dias, et al., 2013). All these contribute to cellular injury in PD.
Sources of Oxidative Stress in Parkinson’s disease
Dopamine metabolism
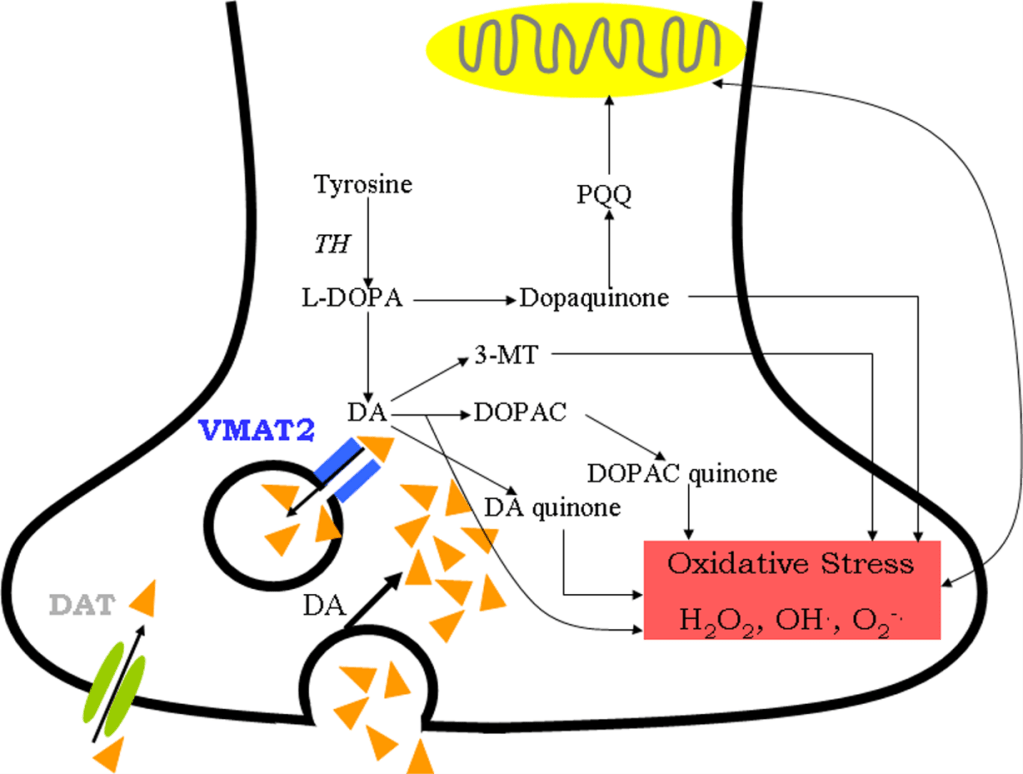
Figure3: Metabolism of cytosolic dopamine produces dopamine quinones that cause oxidative stress in Parkinson’s disease (Qi, et al., 2008).
Dopamine, an unstable neurotransmitter, is normally found in neuron vesicles. But when it is found in the cytoplasm (Hwang, 2013), it undergoes spontaneous auto-oxidation via monoamine oxidase B(MAO-B)-mediated degradation. Vesicular monoamine transporter 2 and dopamine transporter(DAT) helps in recapturing cytosolic dopamine into the vesicles. However, a decline in DAT is seen in patients with PD due to aging and oxidation (Kumar, et al., 2012). Hence, the cytosolic dopamine becomes a source of OxiStres (Hwang, 2013) in PD by synthesizing dopamine quinone(Figure3).
Dopamine quinone can oxidise many proteins like α-synuclein, DJ-1, DAT, parkin, tyrosine hydroxylase(TH)(rate-limiting enzyme in dopamine synthesis), etc. They can also cause mitochondrial dysfunction(by modifying Complex I and II) and proteasome inhibition. Final product of dopamine metabolism is neuromelanin. It can amplify neurodegeneration by triggering neuroinflammation (Hwang, 2013). Therefore, all these can trigger OxiStres, leading to massive cell destruction.
Dysfunctional mitochondria
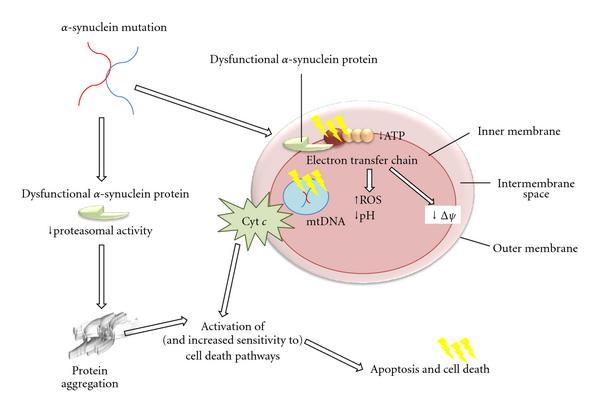
Figure4: Post-events of a dysfunctional mitochondrion leading to oxidative stress followed by apoptosis. The post-events include decreased ATP production, increased ROH production, increased mitochondrial permeability, damaged DNA and release of cytochrome c (Mounsey & Teismann, 2011).
FR are generated normally during OxPhos in mitochondria. Dysfunctional mitochondria(damaged Complex I or II) can occur due to oxidation by dopamine quinones or dysfunctional α-synuclein, parkins, etc (Hwang, 2013). It leads to decreased ATP production, increased OxiStres, altered morphology, impaired calcium buffering, damaged DNA and induction of mitochondrial permeability transition pore (MTP)(decreased mitochondrial-membrane potential:Δψ) (Mounsey & Teismann, 2011). It also oxidises its own lipid called cardiolipin to allow cytochrome c to enter the cytosol and initiate apoptosis. (Hwang, 2013)(Figure4).
Accumulation of iron
Iron in brain supports and catalyses myelination and neurotransmitter synthesis. They are normally stored in ferritins of glial cells (Dias, et al., 2013). A recent study about PD found that despite having normal ferritin levels, iron concentrations in dopaminergic neurons are higher. This is due to iron homeostasis dysregulation (Medeiros, et al., 2016). Hence, accumulation of excessive free iron occurs, causing OxiStres. Free iron reacts with O2●– and H2O2 to give rise to OH● through Haber-Weiss reaction(Figure5), which leads to damaged biomolecules (Liang, et al., 2013).
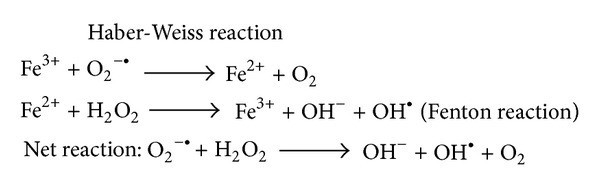
Figure5: Accumulation of excess iron can cause increased production of hydroxyl free radical via Haber-Weiss reaction, which would lead to oxidative (Liang, et al., 2013).
Genetic mutations
The genetic mutations associated with increased OxiStres in PD are listed below:
PTEN-induced putative kinase1(PINK1): Its kinase function helps in providing neuroprotection. If mutated, neurons will become highly vulnerable to MPP+(neurotoxin) and H2O2 and lead to cell injury (S. Gandhi, 2006)(Figure6).
Parkin: It is an E3 ubiquitin ligase, which is important for dopaminergic neuron survival. It helps in tackling OxiStres by translocating itself to depolarized mitochondria to ubiquitinate multiple proteins that would be later degraded by ubiquitin-proteasome system(UPS) (Song, et al., 2017). If mutated, chance for OxiStres increases (Dias, et al., 2013)(Figure6).
UPS: It forms the central pathway for degradation of damaged proteins. Also, it aids in detoxifying OxiStres by producing amino acids that acts as ROS scavengers. Mutation in, parkin or ubiquitin carboxy-terminal hydroxylase L1(UCH-L1), parts of UPS, can aid to loss of dopaminergic neurons in PD(Figure6).
DJ-1: It is a neuroprotective protein that prevents oxidation(enhances GSH levels), apoptosis and inflammation. If mutated, it can cause UPS damage, ER stress and calcium influx (Dias, et al., 2013)(Figure6).
LRRK2: It kinase activity (Dias, et al., 2013) regulates immune system and its inflammatory responses. Its levels are highly increased in patients with PD such that it can cause mitochondrial fragmentation, causing increased ROS production (Cook, et al., 2017)(Figure6).
α-synuclein: It regulates the synaptic plasticity and neurotransmission of dopamine. When mutated, it loses its function and forms aggregations in brain called as Lewy bodies. Lewy bodies are an indicator of neurodegeneration and OxiStres. Its mutated proteins can cause oxidation of other molecules like Complex I, etc, thereby enhances OxiStres (Dias, et al., 2013)(Figure6).
ATP13A2: It is a lysosomal ATPase. Any mutations in this gene can cause malfunction of lysosome and autophagy mechanism (Bento, et al., 2016)(Figure6).
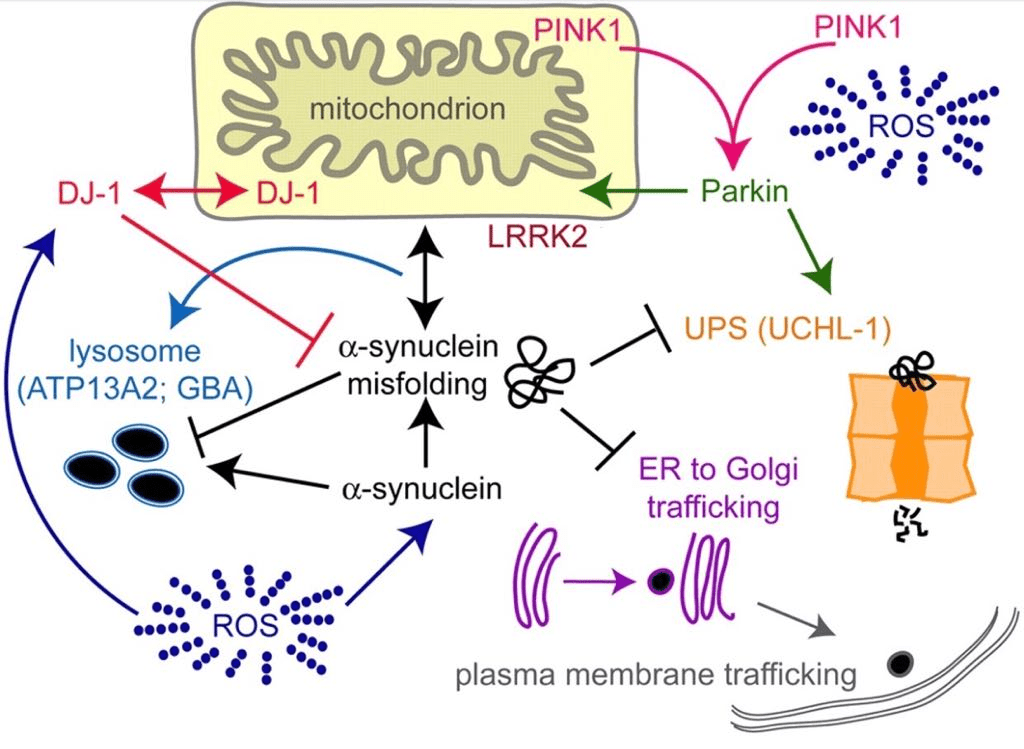
Figure6: Genetic mutations in PINK1, parkin, UPS(UCHL-1), DJ-1, LRRK2, α-synuclein and ATP13A2 are associated with Parkinson’s disease that lead to oxidative stress (Caldwell & Caldwell, 2008).
Neuroinflammation
OxiStres, neurotoxins, neuromelanin, etc can trigger neuroinflammation (Hwang, 2013) by recruiting T-cells, astrocytes and microglia. Their main role is to protect the neurons through production of inflammatory cytokines and ROS. However, in patients with PD, neuroinflammation exacerbates the situation of OxiStres (Taylor, et al., 2013). As microglia is strongly populated in the midbrain, the strength of neuroinflammation will cause detrimental effects to the dopaminergic neurons that are normally susceptible to OxiStres (Dias, et al., 2013)(Figure7).
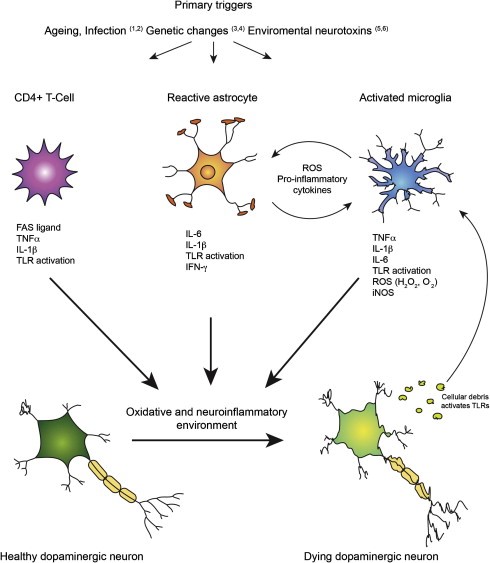
Figure7: Under oxidative stress, neuroinflammation is triggered by T-cells, microglia and astrocyte to promote oxidative stress by releasing more free radicals into the microenvironment of the neurons thereby imparting detrimental effects on them (Taylor, et al., 2013).
Consequences of Oxidative Stress in Parkinson’s disease
Oxidation of proteins and DNA
Oxidative damage to proteins due to ROS can occur via three ways. Firstly, modifications of amino acids like methionine, histidine, arginine and cysteine. Secondly, peptide cleavage by ROS. Lastly, reactions between proteins and lipid peroxidation products. Protein oxidation is identified, if methionine sulfoxide, protein carbonyls, protein peroxides or other amino acid modifications are present (Lobo, et al., 2010). They are accumulated as protein aggregates in the tissue as UPS function is impaired under OxiStres in PD. Protein oxidation is evident in PD in the form of Lewy bodies, which contain proteins like α-synuclein (Dias, et al., 2013). However, there are oxidized proteins like SOD1, which are not found in the Lewy bodies (Halliwell & Gutteridge, 2007).
DNA and RNA are susceptible to oxidative damage by FR. Mostly, the mitochondrial DNA is affected by ROS. Because of which, mutated gene expression will cause dysfunctional mitochondria that have decreased ATP production (Lobo, et al., 2010). The oxidised products can again cause further OxiStres by acting as a source of ROS. A marker for DNA damage in PD is 8-hydroxy-2’-deoxyguanosine ( Adibhatla & Hatcher, 2007).
Lipid peroxidation

Figure8: Sequential steps in lipid peroxidation (MA, et al., 2015).
Lipid peroxidation occurs due to reaction between an ROS and polyunsaturated fatty acid(PUFA), which is present in plasma membrane. The ROS abstracts hydrogen from the PUFA to produce a lipid radical, which would then initiate chain reactions to form diene conjugate, peroxyl radical, lipid hydroperoxide sequentially(Figure8). Markers for lipid peroxidation in PD include malondialdehyde(Figure8), isoprostanes (MA, et al., 2015) and 4-Hydroxyl-2-nonenal(HNE). HNE contributes to GSH decline and apoptosis of neurons (Dias, et al., 2013). The PUFA can also bind to α-synuclein (Galvagnion, 2017) after dislodging from the cell membrane with the help of phospholipases A2 (Sun, et al., 2014). The presence of lipid derivatives in Lewy bodies, which mainly contain α-synuclein, has been verified. Heavy lipid peroxidation is seen in PD because of the brain’s second highest lipid concentration after adipose tissue. As lipids are the main component of plasma membrane, lipid peroxidation can impair permeability and integrity of membranes (Rao, et al., 2011), which may lead to apoptosis.
Altered Calcium influx
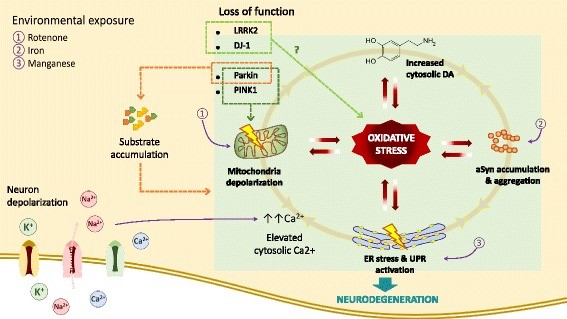
Figure9: Sources of oxidative stress like dopamine metabolism, dysfunctional mitochondria, and genetic mutations can cause calcium influx in the neurons (Puspita, et al., 2017).
Calcium regulation is altered under OxiStres, which leads to altered cell signalling. Majority of the neurons use sodium channels than calcium channels to maintain action potential. However, SNc and SNr mainly use L-type calcium channels to regulate pacemaking. They also have great demands for OxPhos. Because of these features, neurons at substantia nigra are more susceptible to OxiStres and lead to neurodegeneration. The chief organelles responsible for calcium influx is mitochondria and ER (Kumar, et al., 2012). Under oxidative stress, those organelles become dysfunctional and are under stress, thereby aids to enhance calcium influx(Figure9).
Cell death
Under OxiStres, macromolecules are oxidised and are left malfunctioned. Since, they no longer continue their function, the cell would eventually die. This is what happens in PD also. As each organelle and its function are affected, more unwanted waste and toxic substances start accumulating at the site of injury. Finally, the situation will go beyond the threshold, where neurodegeneration i.e. neuron death will take place.
Defence system
The defence system of brain involves antioxidants and neurotrophic factors.
Antioxidants
They are electron rich and therefore, can stabilise FR by donating its electrons without getting unstable. However, in patients with PD, the quantity of antioxidants is not enough to combat OxiStres. Therefore, weak antioxidant activity is responsible for progression of PD. Antioxidants can be enzymatic(GSH systems, catalase and SOD) and non-enzymatic(tocopherols and tocotrienols, GSH, ascorbic acid and melatonin).
SOD: They are intracellular antioxidants that catalyse the reaction of O2●- into O2 and H2O2 with the help of Cu/Zn, Fe or Mn.
GSH systems: They are intracellular antioxidants involving GSH, GSH reductase, GSH peroxidases and GSH S-transferases. They are scavengers of hydrogen peroxide and lipid peroxides.
Ascorbic acid: They are dietary antioxidants, also called as Vitamin C, that act as reducing agents to scavenge ROS.
Melatonin: They are powerful intracellular antioxidants that can cross the blood-brain barrier. They don’t undergo redox reactions. If it gets oxidised, it cannot revert. Hence, they are also called as suicide antioxidants.
Tocopherols and tocotrienols: They are dietary antioxidants, also called as Vitamin E, that are involved in protection of plasma membrane against the attack of FR (Lobo, et al., 2010).
Neurotrophic factors(NTF)
NTF produced in the brain include glial cell line-derived neurotrophic factor(GDNF), mesencephalic astrocyte-derived neurotrophic factor(MANF), neurturin(NRTN), cerebral dopamine neurotrophic factor(CDNF), etc. They function to protect dopaminergic neurons in PD. Knowing their protective ability, clinical research is made on NTF-based therapy for PD. A limitation of this therapy is that it involves invasive techniques (Goswami, et al., 2017).
Conclusion
There are many factors that cause OxiStres in PD. A single factor is enough to trigger ROS production, which can damage other organelles and biomolecules to either make them a source of oxidative stress or prevent their function from defending against oxidative stress. All the factors and consequences together contribute to cellular injury in PD. Due ROS’ reactive nature, it is very hard to prevent any disease caused by ROS. However, once the disease is diagnosed as quickly as possible, treatments on alleviating the symptoms and cellular damage can be provided. However, one thing to be noted is that PD is not curable (Connolly & Lang, 2014). Hence, more research is required to comprehend effectively on how to prevent or cure PD.
References
Adibhatla, R. M. & Hatcher, . J. F., 2007. Role of Lipids in Brain Injury and Diseases. Future lipidology, [Online]. 2(4), pp. 403-422. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2174836/pdf/nihms30796.pdf [Accessed 4 June 2018].
Bento, C. . F., Ashkenazi, A., Jimenez-Sanchez, . M. & Rubinsztein, D. . C., 2016. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nature Communications,[Online]. 7(1). Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4906231/ [Accessed 4 June 2018].
Caldwell, G. . A. & Caldwell, K. A., 2008. Traversing a wormhole to combat Parkinson’s disease. Disease Models & Mechanisms,[Online]. 1(1), pp. 32-36.Available at:
http://dmm.biologists.org/content/1/1/32 [Accessed 4 June 2018].
Connolly, B. . S. & Lang, . A. E., 2014. Pharmacological Treatment of Parkinson DiseaseA Review. JAMA, [Online]. 311(16), p. 1670–1683. Available at:
http://dosequis.colorado.edu/Courses/BrainWeb/papers/PARK1.pdf [Accessed June 2018].
Cook, D. A. et al., 2017. LRRK2 levels in immune cells are increased in Parkinson’s disease. Parkinson’s Disease,[Online]. 3(11). Available at:
https://www.nature.com/articles/s41531-017-0010-8 [Accessed 4 June 2018].
Dias, V., Junn, E. & Mouradian, M. M., 2013. The Role of Oxidative Stress in Parkinson’s Disease. Journal of Parkinson’s disease,[Online]. 3(4), pp. 461-491. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4135313/ [Accessed 4 June 2018].
Galvagnion, C., 2017. The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson’s Disease. Journal of Parkinson’s Disease, [Online]. 7(3), pp. 433-450. Available at:
https://content.iospress.com/articles/journal-of-parkinsons-disease/jpd171103 [Accessed 4 June 2018].
Goswami, P., Joshi, N. & Singh, S., 2017. Neurodegenerative signaling factors and mechanisms in Parkinson’s pathology. Toxicology in Vitro,[Online]. 43(1), pp. 104-112. Available at:
https://www.sciencedirect.com/science/article/pii/S0887233317301571 [Accessed 4 June 2018].
Halliwell, B. & Gutteridge, J. M., 2007. Free radicals in biology and medicine. 4 ed. New York: Oxford University Press.
Hwang, O., 2013. Role of Oxidative Stress in Parkinson’s Disease. Experimental neurobiology,[Online]. 22(1), pp. 11-17. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3620453/pdf/en-22-11.pdf [Accessed 4 June 2018].
Jarmuszkiewicz, W. et al., 2015. Temperature controls oxidative phosphorylation and reactive oxygen species production through uncoupling in rat skeletal muscle mitochondria. Free Radical Biology and Medicine,[Online]. 83(1), pp. 12-20.Available at:
https://www.sciencedirect.com/science/article/pii/S0891584915000751?via%3Dihub [Accessed 4 June 2018].
Kumar, H. et al., 2012. The Role of Free Radicals in the Aging Brain and Parkinson’s Disease: Convergence and Parallelism. International Journal of Molecular Sciences, [Online]. 13(8), pp. 10478-10504. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3431873/pdf/ijms-13-10478.pdf [Accessed 4 June 2018].
Leisman, G., Melillo, R. & R, F., 2013. The basal ganglia: motor and cognitive relationships in a clinical neurobehavioral context. Reviews in the Neurosciences,[Online]. 24(1), pp. 9-25. Available at:
https://www.researchgate.net/publication/233930154_The_basal_ganglia_Motor_and_cognitive_relationships_in_a_clinical_neurobehavioral_context [Accessed 4 June 2018].
Liang, S.-S.et al., 2013. Online Monitoring Oxidative Products and Metabolites of Nicotine by Free Radicals Generation with Fenton Reaction in Tandem Mass Spectrometry. The Scientific World Journal, [Online]. 2013(1). Available at:
https://www.hindawi.com/journals/tswj/2013/189162/ [Accessed 4 June 2018].
Lobo, V., Patil, . A., Phatak, . A. & Chandra, . N., 2010. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacognosy Review,[Online]. 4(8), p. 118–126. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3249911/ [Accessed 4 June 2018].
MA, G., G, . R. & N, . D., 2015. Marine Carotenoids against Oxidative Stress: Effects on Human Health.. Marine drugs,[Online]. 13(10), pp. 6226-6246. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4626686/ [Accessed 4 June 2018].
Magrinelli,, F. et al., 2016. Pathophysiology of Motor Dysfunction in Parkinson’s Disease as the Rationale for Drug Treatment and Rehabilitation. Parkinson’s disease,[Online]. 2016(1). Available at:
https://www.hindawi.com/journals/pd/2016/9832839/ [Accessed 4 June 2018].
Manoharan, S. et al., 2016. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxidative medicine and Longevity Medicine,[Online]. 2016(1). Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5223034/ [Accessed 4 June 2018].
Medeiros, M. S. et al., 2016. Iron and Oxidative Stress in Parkinson’s Disease: An Observational Study of Injury Biomarkers. PLoS One, [Online]. 11(1). Available at:
http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0146129 [Accessed 4 June 2018].
Mounsey, R. B. & Teismann, P., 2011. Mitochondrial Dysfunction in Parkinson’s Disease: Pathogenesis and Neuroprotection. Parkinson’s disease,[Online]. 2011(1), p. 18. Available at:
https://www.hindawi.com/journals/pd/2011/617472/ [Accessed 4 June 2018].
Nambu, A., 2009. Basal Ganglia: Physiological Circuits. In: Encyclopedia of Neuroscience. s.l.:Elsevier, [Online]. pp. 111-117. Available at:
https://www.researchgate.net/…/BASAL+GANGLIA+SUMMARY+ARTICLE.pdf [Accessed 4 June 2018].
Nelson, A. & Kreitzer, A., 2014. Reassessing Models of Basal Ganglia Function and Dysfunction. Annual review of neuroscience, [Online]. 37(1), pp. 117-135. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4416475/ [Accessed 4 June 2018].
Obeso, J. A. et al., 2014. The expanding universe of disorders of the basal ganglia. The Lancet,[Online]. 384(9942), pp. 523-531. Available at:
https://www.thelancet.com/pdfs/journals/lancet/PIIS0140-6736(13)62418-6.pdf [Accessed 4 June 2018].
Potkonjak, D. & Cox, B., 1969 . The relationship between tremor and change in brain acetylcholine concentration produced by injection of tremorine or oxotremorine in the rat. British Journal of Pharmacology, [Online]. 35(2), pp. 295-303. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1703230/pdf/brjpharm00574-0091.pdf [Accessed 4 June 2018].
Puspita, L., Chung, S. Y. & Shim, J.-w., 2017. Oxidative stress and cellular pathologies in Parkinson’s disease. Molecular Brain,[Online]. 10(53). Available at:
https://molecularbrain.biomedcentral.com/articles/10.1186/s13041-017-0340-9 [Accessed 4 June 2018].
Qi, Z., Miller, G. W. & Voit, E. . O., 2008. Computational Systems Analysis of Dopamine Metabolism. Plos One,[Online]. 3(6), p. e2444. Available at:
http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0002444 [Accessed 4 June 2018].
Rao, S. P., Kalva, S., Yerramilli, A. & Mamidi, S., 2011. Free radicals and tissue damage: role of antioxidants. Free radicals and antioxidants,[Online]. 1(4), pp. 2-7. Available at:
http://www.phcogfirst.com/sites/default/files/AX_1_4_2.pdf [Accessed 4 June 2018].
Roberts, R. . F., Wade-Martins, . R. & Alegre-Abarrategui, J., 2015. Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain, [Online]. 138(6), pp. 1642-1657. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4614141/ [Accessed 4 June 2018].
S. Gandhi, M. M. K. M. L. S. D. G. H. P. M. A.-S. I. H. S. H. M. G. L. P. A. J. L. D. S. L. J. L. H. N. W. W. T. R., 2006. PINK1 protein in normal human brain and Parkinson’s disease. Brain,[Online]. 129(7), pp. 1720-1731.Available at:
https://academic.oup.com/brain/article/129/7/1720/303945 [Accessed 4 June 2018].
Song, L., McMackin, M., Nguyen, A. & Cortopassi, G., 2017. Parkin deficiency accelerates consequences of mitochondrial DNA deletions and Parkinsonism. Neurobiology of Disease,[Online]. 100(1), pp. 30-38. Available at:
https://www.sciencedirect.com/science/article/pii/S096999611630314X [Accessed 4 June 2018].
Sun, G. Y. et al., 2014. Role of cytosolic phospholipase A2 in oxidative and inflammatory signaling pathways in different cell types in the central nervous system. Molecular neurobiology,[Online]. 50(1), pp. 6-14. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4147031/ [Accessed 4 June 2018].
Taylor, J. M., Main, B. S. & Crack, P. . J., 2013. Neuroinflammation and oxidative stress: Co-conspirators in the pathology of Parkinson’s disease. Neurochemistry International,[Online]. 62(5), pp. 803-819.Available at:
https://www.sciencedirect.com/science/article/pii/S0197018612004147?via%3Dihub [Accessed 4 June 2018].
